この度、技術情報協会が発行する製薬・原材料・医療機器企業のための新しい教育・実務雑誌『月刊PHARMSTAGE』8月号(8月15日発行予定)において、弊社の薬事・品質保証を担当する村上の記事が掲載されます。
米国におけるコンビネーションプロダクト開発における規制・ガイドライン
(The regulation and guideline for the combination product development in US)
1 コンビネーションプロダクト
1.1 コンビネーションプロダクトの定義及び製品例
コンビネーションプロダクトとは、医薬品、医療機器及び生物学的製剤等を組み合わせた診断用又は治療用の製品をいう。コンビネーションプロダクトは、製品の開発から今日に至るまで世界各国でさまざまな製品が臨床使用されてきたが、世界で統一された定義が明確には定められている訳でない。近年、本邦ではコンビネーションプロダクトの現状を踏まえ、これらの規制について議論がなされているが、コンビネーションプロダクトの開発等で先行する欧米に倣い、コンビネーションプロダクトの定義や規制を運用している。なお、米国では、21 CFR 3.2 (e) にてコンビネーションプロダクトが定義されている1)。
(1) 米国におけるコンビネーションプロダクトの定義
21 CFR 3.2 (e) で定義されているコンビネーションプロダクトは以下のとおりである。
① 2つ以上の規制対象となるコンポーネントを物理的、化学的、あるいは他の方法で組み合わせるか混合して一つの製品としたもの
(a) 医薬品/医療機器
(b) 生物学的製剤/医療機器
(c) 医薬品/生物学的製剤
(d) 医薬品/医療機器/生物学的製剤
② 2つ以上の個別製品(医薬品/医療機器、医療機器/生物学的製剤又は生物学的製剤/医療機器)が、ユニットあるいは単一包装された製品
③ 医薬品、医療機器又は生物学的製剤が個別包装されており、治療計画や製品ラベルに従い、それぞれの使用目的等を達成することを意図して一緒に使用する製品
④ 研究用の医薬品、医療機器又は生物学的製剤が個別包装されており、治療計画や製品ラベルに従い、それぞれの使用目的等を達成することを意図して一緒に使用する製品
(2) 米国におけるコンビネーションプロダクトの製品例
21 CFR 3.2 (e) に該当するコンビネーションプロダクトの例としては以下のものが挙げられる(これに限られるものではない)。
①プレフィルドシリンジ(薬品があらかじめ充填された注射器)
② ペン型インスリン注入器
③ 経口吸入デバイス
④ 薬剤溶出型ステント
⑤ 薬剤コーティング(含侵)カテーテル
⑥ 薬剤コーティング(ペースメーカー用)リード
⑦ 抗生物質含有の骨セメント
⑧ 殺精子剤を含有したコンドーム
1.2 コンビネーションプロダクトの分類について
コンビネーションプロダクトは、製品の組み合わせの内容、性質や特徴を勘案し、適用される規制や審査部門が指定される。米国では、2002年12月にMedical Device User Fee and Modernization Act of 2002 (MDUFMA) の第204章 で規定され2)、設立されたOffice of Combination Products (OCP) が、コンビネーションプロダクトに係る案件の窓口となり、専門的に問題解決などにあっている。OCPにおける役割は以下のとおりである。
(1) OCPの役割
① 審査部門及び申請者向けにコンビネーションプロダクトに関する見解の集約
② コンビネーションプロダクトの規制やガイダンス文書の作成
③ 製品の分類(非コンビネーションプロダクト(医薬品、医療機器、生物学的製剤)又はコンビネーションプロダクトなど)、並びに管轄する審査部門の指定
④ 審査部門間の調整を行い、適時適切な市販前審査の確保
⑤ コンビネーションプロダクトの適切かつ継続性のある市販後規制の確保
⑥ コンビネーションプロダクトの市販前審査スケジュールに関する問題(係争)の解決
⑦ コンビネーションプロダクトに係るガイダンス文書等の更新
⑧ 年次業務報告書の作成
(2) 審査部門の指定
上記のとおり、OCPではコンビネーションプロダクトの特性を踏まえ、審査部門を生物製剤評価センター(CBER)、医薬品評価センター(CDER)、医療機器・放射線保健センター(CDRH)のいずれかに指定する。この指定にあたり、申請者はOCPに公式又は非公式に接触する必要があり、何れの方法によっても管轄する審査部門の指定を受ける必要がある。公式な手続きとしては、 21 CFR Part 3で定められている Request for Designation Document (RFD) を作成し、OCPに提出する必要がある3)。
1.3 RFDプロセス
RFDは、コンビネーションプロダクト又は非コンビネーションプロダクトの規制上の識別や分類、コンビネーションプロダクトである場合、ない場合を問わず、どの部門で審査を受けることになるかを確認する必要がある場合に提出されるものである。コンビネーションプロダクトに該当すると考えられる品目は必ずこのRFDを提出しなければならないというものではなく、開発製品の分類に疑義がある場合に提出が求められるものである。OCPは、申請者からRFDが提出されると60日以内に担当審査部門を指定しなければならない。OCPでは、Primary Mode of Action (PMOA) (治療や診断にとって最も重要な部分)に基づき、対象となる規制、審査部門を指定することになる。
表1は、米国においてRFDが提出された後に、コンビネーションプロダクトあるいは非コンビネーションプロダクトに指定された品目数の推移を示したものである。いずれの年においても、コンビネーションプロダクトと判断された品目の方が多いが、2013年にコンビネーションプロダクトと判断された品目数は、直近の5年でコンビネーションプロダクトに指定された品目数の平均値に比べて26% 減少している。一方、2013年に非コンビネーションプロダクトに指定された品目数は直近5年の品目数と比較して27% 増加している。
【表1. OCPによるRFDの判定結果(年別の品目数の推移)】
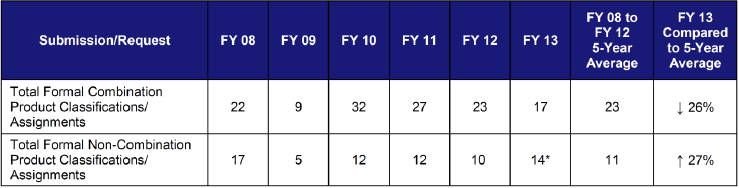
表2は、2013年にOCPに提出されたRFD (31品目)のうち、コンビネーションプロダクトに指定された17品目の分類に関する内訳であるが、その大多数が医薬品と医療機器のコンビネーションプロダクトであった。
【表2. コンビネーションプロダクトの分類(17品目/2013年)】
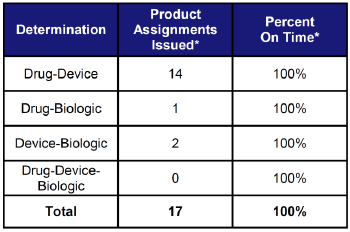
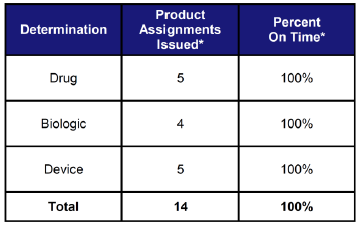
表3は、非コンビネーションプロダクトと判断された14品目の内訳である。医薬品、医療機器及び生物学的製剤に指定された品目数はほぼ同数であった。
なお、2013年のOCPへ非公式(RFDによるものではない)に接触があった件数は233件であるが、2012年と比較して件数は13% 減少したと報告されている4)。なお、2012年までの過去5年の件数は、それぞれ268件 (2012)、265件 (2011)、303件 (2010)、256件 (2009)、190件 (2008) であり、2008年以来の低水準であった5~9)。
【表3. 非コンビネーションプロダクトの分類(14品目/2013年)】
2 規制及びガイドライン
米国では、1990年にコンビネーションプロダクトの概念が導入され、当該製品の管轄を明確化する目的でFederal Food, Drug and Cosmetic Act (the Act) が改正された。翌年、CBER、CDER及びCDRHの3部門により、Intercenter Agreements (ICAs) が締結され、コンビネーションプロダクトの管轄や共同審査について合意されている。2002年にはコンビネーションプロダクトに関する処置、取り扱い、公式の協議や共同審査についてトラッキングするため、FDAスタッフ向けの内部文書が発行されている。翌年に一部を改訂し、暫定的な形ではあったがこの取り組みが実施され、2005年にはPMOAの最終規則が決定された。以後、継続的にガイダンス文書が発行されている。2011年4月にRFDプロセスが制定されると、同年6月には製品の分類に関して、特に医薬品又は医療機器に該当する可能性のある製品に特化する形でガイダンスが発行された。表2にて記載されているが、従来、コンビネーションプロダクトはそのほとんどが医薬品又は医療機器に分類されることが多く、それに対応したものである。また、本年1月にはコンビネーションプロダクトに関するcGMPのガイダンスが発行されている。cGMPは、2013年1月に21 CFR part 4にて制定され、医薬品、医療機器、生物学的製剤及びヒト細胞、組織又は細胞・組織由来製品について定めたものであるが、コンビネーションプロダクトに関しては定められていなかった。このガイダンスは、これらの製品に加えて、コンビネーションプロダクトのルールを明確に定めたものである。
また、個別の製品に関するガイドラインも制定されてきており、
2013年4月には、ガラス製シリンジを対象とした技術情報、同年6月には、ペン型、ジェット型などの注入器に関する技術的考慮事項についてガイダンスが発行されている。
現在、世界各国でコンビネーションプロダクトの取り扱いについてさまざまな議論がなされているが、本邦でも厚生労働省、独立行政法人医薬品医療機器総合機構(PMDA)が、各国の規制当局と相互に協議し、規制の在り方や課題への取り組みについて継続的に検討しているところである。その意味で米国をはじめ、海外のコンビネーションプロダクトの規制について学ぶことは重要になる。
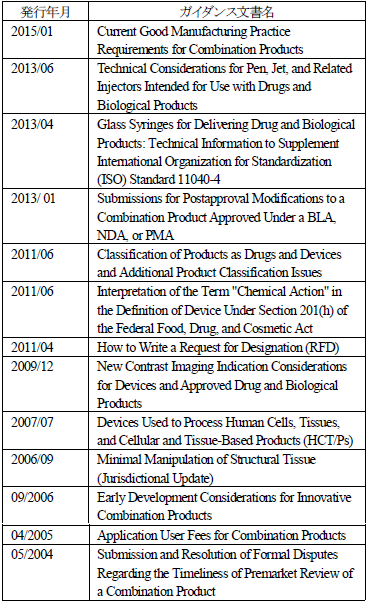
最後に、FDAから発行されているガイダンス文書を以下に紹介する。
Reference:
1. Combination product – section 503(g) of the Federal Food, Drug, and Cosmetic Act [21 U.S.C 353 (g)] and 21 CFR Part 3.2(e)
2. The Office of Combination Products was established on December 24, 2002 as required by SEC. 204 of the Medical Device User Fee and Modernization Act of 2002 (MDUFMA).
3. 21 CFR Part 3 – the regulations pertaining to the assignment of products to FDA components for review of premarket applications
4. FY 2013 PERFORMANCE REPORT TO CONGRESS for the Office of Combination Products as required by the Medical Device User Fee and Modernization Act of 2002
5. FY 2012 PERFORMANCE REPORT TO CONGRESS for the Office of Combination Products as required by the Medical Device User Fee and Modernization Act of 2002
6. FY 2011 PERFORMANCE REPORT TO CONGRESS for the Office of Combination Products as required by the Medical Device User Fee and Modernization Act of 2002
7. FY 2010 PERFORMANCE REPORT TO CONGRESS for the Office of Combination Products as required by the Medical Device User Fee and Modernization Act of 2002
8. FY 2009 PERFORMANCE REPORT TO CONGRESS for the Office of Combination Products as required by the Medical Device User Fee and Modernization Act of 2002
9. FY 2008 PERFORMANCE REPORT TO CONGRESS for the Office of Combination Products as required by the Medical Device User Fee and Modernization Act of 2002
『月刊PHARMSTAGE』8月号は書店または技術情報協会ホームページよりご購入いただけます
技術情報協会ホームページ:http://www.gijutu.co.jp/doc/magazine_pharm%20stage.htm

